

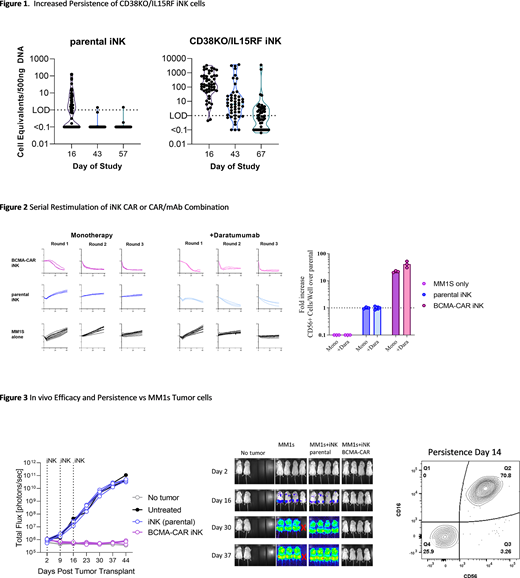
Multiple redundancy within the spectrum of an immune response is required to prevent antigen escape or adaptation of the targeted population to host defenses. As adoptive cell therapies continue to evolve, multi-modal engineering of effector cells offers the prospect of tackling increasingly complex disease settings such as multiple myeloma (MM), where targeting of a single tumor associated antigen is frequently confounded by antigen shedding and escape variation resulting in the inability to develop a curative therapy. There are multiple advantages in expanding treatment options beyond autologous primary T and NK cells, including the use of induced pluripotent stem cells (iPSC) to derive effector cells that can be uniformly manufactured at scale from renewable starting cellular material and where precision genetic engineering can be achieved at the clonal level which can be applied sequentially in order to build multiple specificities and functional modalities. To create a platform targeted toward MM, a multiplexed edited base iPSC-derived NK (iNK) cell configuration consisting of a CD38 KO iPSC modified to overexpress a recombinant IL-15 signaling complex (IL15RF) for autonomous persistence and a functionally enhanced high-affinity, non-cleavable CD16 (hnCD16) was developed. Introduction of IL15RF enabled expansion of iNK cells without additional exogenous cytokine support during the manufacturing process and greatly improved functional persistence of iNK cells both in vitro and in various xenograft mouse models (Figure 1).
To target MM in a broad and comprehensive manner, we tested our novel BCMA-CAR in combination with different myeloma targeted antibodies. In combination with hnCD16, co-expression of BCMA-CAR and IL15RF culminates in an iNK cell therapeutic, termed FT576, capable of multiantigen-specificity through combinatorial use of CAR and hnCD16 with monoclonal antibodies to tackle antigen escape. Chimerization of an anti-BCMA scFv shown to elicit higher affinity onto the CAR platform produced specific in vitro recognition of BCMA+ myeloma cells in short-term and long-term NK cell cytotoxicity assays. Specificity of the BCMA-CAR was demonstrated using NALM6 overexpressing BCMA using a short range 4H caspase assay (NALM6_BCMA EC50 14.4, NALM6wt EC50 39.1, p*<0.0001).
Utilizing a long range clearance assay, serial restimulation by repeated rounds of exposure to fresh MM1S MM target cells was tested, showing remarkable persistence and antigen-mediated expansion of CAR function in isolation or combined with antibody through 3 rounds of stimulation in the absence of exogenous cytokine support (Figure 2). Continuous long-range clearance assays demonstrated levels of BCMA targeting activity of FT576 alone was equivalent to primary BCMA-targeted CAR-T cells against a panel of BCMA+ target cells. Utilizing hnCD16, BCMA-CAR was tested in combination with anti-CD38 (daratumumab), anti-SLAMF7 (elotuzumab), or anti-CD19, showing synergistic increase in tumor targeting through various tumor associated antigens (TAAs). Polyfunctionality of FT576 stimulated either through CAR or ADCC was similarly measured by both Isoplexis and single cell RNA sequencing. Specificity for plasma cells was confirmed using primary bone marrow samples from either healthy donors or patients.
In animal models, as a monotherapy, FT576 achieved sustained tumor control against disseminated MM1s with persistence profile suggestive of antigen mediated expansion (Figure 3). In combination with daratumumab, FT576 was able to achieve complete clearance of MM1S. Combination with other monoclonal antibodies displayed a similar response demonstrating the unique ability of FT576 to be directed to target multiple TAAs. Together, these studies demonstrate the versatility of FT576 as a highly effective multi-antigen targeting and cost-effective off-the-shelf BCMA-CAR iNK cell product and supports the rational for a first-of-kind Phase I Study as a monotherapy or in combination with therapeutic mAbs targeted to MM-associated surface antigens, driving a path towards a curative therapeutic in MM.
Goodridge:Fate Therapeutics, Inc: Current Employment. Bjordahl:Fate Therapeutics: Current Employment. Mahmood:Fate Therapeutics, Inc: Current Employment. Reiser:FATE THERAPEUTICS: Current Employment. Gaidarova:Fate Therapeutics, Inc: Current Employment. Blum:Fate Therapeutics: Current Employment. Cichocki:Fate Therapeutics, Inc: Consultancy, Patents & Royalties, Research Funding. Chu:Fate Therapeutics, Inc: Current Employment. Bonello:Fate Therapeutics, Inc: Current Employment. Lee:Fate Therapeutics, Inc.: Current Employment. Groff:Fate Therapeutics, Inc: Current Employment. Meza:Fate Therapeutics, Inc: Current Employment. Chu:Roche Holding AG: Current equity holder in publicly-traded company; Fate Therapeutics, Inc.: Current Employment, Current equity holder in publicly-traded company. Walcheck:Fate Therapeutics: Consultancy, Research Funding. Malmberg:Vycellix: Membership on an entity's Board of Directors or advisory committees; Fate Therapeutics: Consultancy, Patents & Royalties. Miller:Vycellix: Consultancy; Onkimmune: Honoraria, Membership on an entity's Board of Directors or advisory committees; Nektar: Honoraria, Membership on an entity's Board of Directors or advisory committees; Fate Therapeutics, Inc: Consultancy, Patents & Royalties, Research Funding; GT Biopharma: Consultancy, Patents & Royalties, Research Funding. Valamehr:Fate Therapeutics, Inc: Current Employment, Current equity holder in publicly-traded company.

